| Mitochondrial Energetics
CALCIUM ACTIVATION OF OXIDATIVE PHOSPHORYLATION: CHANGES IN ATP SYNTHESIS RATE WITH CLAMPED NADH REDOX STATE.
Paul R. Territo, Stephanie A. French, and Robert S. Balaban
Laboratory of Cardiac Energetics, National Heart Lung and Blood Institute,
Bethesda Maryland 20892 USA
Introduction
Early regulatory models of oxidative phosphorylation (ox-phos) were thought to occur via cytosolic feedback of ATP hydrolysis (1). The net result of such regulation is a decrease in metabolic intermediates (ADP, Pi) and the dissipation of NADH driving force for ATP synthesis. However, work in a large variety of tissues do not support this theory (2-6), and instead have demonstrated that ATP hydrolysis and synthesis rates are balanced with workload, therefore minimizing fluctuations in metabolic intermediates. For this type of regulation to work, a cytosolic transducer is required which is capable of stimulating both work (ATP hydrolysis) and ATP synthesis in parallel.
Calcium, and known second messenger of the cell, has been shown to stimulate both mechanical and mitochondrial work (ATP production) in parallel, thus minimizing changes in [ATP] with workload (2,7). Similarly, addition of Ca2+ to isolated mitochondria results in an activation of mitochondrial NADH and ATP production rates in parallel, via the calcium sensitive dehydrogenases (CaDH) (8,9) and the F0/F1ATPase (9,10), respectively. Combined, these data suggest that two parallel networks of metabolic regulation exist, at the cell and within the mitochondria, which are mediated by Ca2+, and could serve to balance metabolic demand with supply, as observed in vivo.
Therefore, we hypothesize that Ca2+ could activate mitochondrial State 3 ATP production rate without changes in NADH redox status, by parallel stimulation of the CaDH and F0/F1ATPase in parallel with cytosolic ATPases, and observed in vivo.
Methods and Materials
Porcine hearts were harvested and retrograde perfused (40C) as described previously (9,11). Mitochondria were isolated (9,11) and cytochrome A (Cyta) content was determined as described in (12). Isolated mitochondria were loaded stoichiometrically with 1 nmol 5(6)-carboxy-2’-7’-dichlorofluoresceindiacitate succinimidyl ester (CF; Molecular Probes, Eugene OR) per nmol Cyta at 40C as described in (8,16). In all cases experiments were run at 1 nmol Cyta.ml-1 running buffer.
ATP production (O2 consumption (mVO2)) and membrane potential (Dy) were determined in a closed system respirometer (370C) described in (9). Dy was estimated from Nernst equilibrium of 4 mM TPP+ across the mitochondrial membrane (9), and detected by a TPP+ ion-selective electrode. In all cases electrode drift and non-specific binding of TPP+ to matrix membranes were corrected as per (13). Simultaneous measures of pyridine dinucleotides redox status were monitored at steady state as described in (9,11). NADH was expressed as the ratio of coefficients (INADH.ICF-1) from least squares linear regressions for each spectra, thereby eliminating 10 and 20 scattering. Spectra obtained at various metabolic states had a high degree of concordance both within (0.9922?0.0004, average n=32) and between (0.9921?0.0003, n=92) studies.
In all cases mitochondria were Ca2+ depleted for 6 min. in running buffer (125mM KCl, 20mM HEPES, 15mM NaCl, 5mM MgCl2, 1mM K2EDTA, 1mM EGTA, 2mM Pi, 0.1mM malate, 4mM TPP+, with 3.4mM Na2ATP added fresh daily, pH 7) prior to start of the experiment. Substrates, ATPase ,and Ca2+ were added as per figure legends. Calculations of free Ca2+ were determined from affinity constants described previously (14).
Experimental Design
 |
To test the hypothesis that Ca2+ is capable of stimulating the CaDH and F0/F1ATPase in parallel with increases in cytosolic work, exogenous ATPase and Ca2+ were added simultaneously to cardiac mitochondria at “resting” metabolic rate, thus simulating an increase in cytosolic work, SR Ca2+ release, maximal Ca2+ stimulated metabolic rate as in vivo.
“Resting” metabolism was obtained with ATPase titration to 2X the State 2 rate. Simultaneously metabolic rate (mVO2), metabolic driving forces for ATP synthesis (NADH and DY) were monitored in the chamber to the left.
|
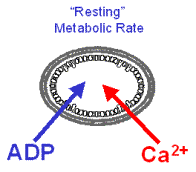 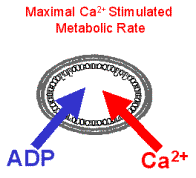 |
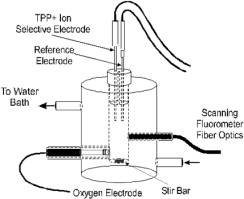
Figure 1.
Schematic diagram of chamber setup used to collect mVO2, NADH and DY simultaneously. The experimental design used in these studies, where mVO2 , NADH, and DY were acquired with time. |
Results
![Steady state plots of mVO2 (A), NADH (B), and DY (C) for mitochondria oxidizing G/M with [Ca2+].](images/mito-calcium-activation10.gif)
Figure 3.
Steady state plots of mVO2 (A), NADH (B), and DY (C) for mitochondria oxidizing G/M with [Ca2+]. Buffers and Ca2+ depletion were as indicated in methods, while substrate, ATPase, and Ca2+ additions were as indicated in Figure 1. Free Ca2+ was calculated as per (14) based on published affinity constants. “Rest’ is defined as 2X State 2 metabolic rate In all cases data are means ? SEM, asterisks (?) indicate significant differences from “rest”, while double crosses (‡) indicate significant difference from nominally zero [Ca2+] (p?0.05; dependent variable t-test).
Proposed Parallel Activation of Cytosolic Work and Mitochondrial Respiration by Ca2+
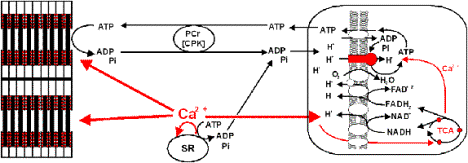
Proposed Mechanisms of Clamped NADH Redox State with Ca2+ and ATPase.
![Ca2+ Activates CaDH and Fo/F1ATPase in parallel with Cytosolic ATPases, resulting in a net balance of [NADH],[ADP],and [ATP].](images/mito-calcium-heart.gif)
Ca2+ Activates CaDH and F0/F1ATPase in parallel with Cytosolic ATPases, resulting in a net balance of [NADH],[ADP],and [ATP].
Conclusions
Ca2+ activated mVO2, NADH, and DY in a dose dependent manor over the [Ca2+] range from 0nM to 535nM.
Maximal State 3 mVO2 was obtained at 535nM Ca2+, and increased by ~1.5 fold over control (nominally zero Ca2+).
Steady state NADH levels, were also maximal at 535nM Ca2+, and increased by ~1.4 fold over control levels.
In contrast, DY decreased by ~1.1 fold from 0nM Ca2+, when maximally stimulated with 535nM Ca2+.
Clamped NADH redox state was achieved at 172nM free Ca2+ (K0.5) plus ATPase, resulting in a >3 fold increase in mVO2 , maintenance of NADH redox state, and an ~1.1X decreased in DY when compared to “resting” conditions.
Combined these data suggest that Ca2+ activation of ox-phos is capable of increasing metabolic flux by several fold, while maintaining metabolic intermediates (NADH, ADP, Pi and ATP) and resulting driving force for ATP synthesis, and may represent a mechanism by which metabolic supply is increased in parallel with elevated demand, as seen in vivo.
References
B. Chance, G. Williams, J. Biol. Chem. 217, 383-393 (1955).
L. A. Katz, J. A. Swain, M. A. Portman, R. S. Balaban, Am J Physiol 256, H265-74 (1989).
J. A. Detre, D. S. Williams, A. P. Koretsky, NMR Biomed 3, 272-8 (1990).
A. P. Koretsky, Physiol Rev 75, 667-88 (1995).
S. D. Wolff, C. Eng, R. S. Balaban, Am J Physiol 255, F581-9 (1988).
C. J. Wingard, R. J. Paul, R. A. Murphy, J Physiol (Lond) 501, 213-23 (1997).
R. S. Balaban, H. L. Kantor, et al., Science 232: 1121-3 (1986).
R. M. Denton, J. G. McCormack, Annu Rev Physiol 52, 451-66 (1990).
P. R. Territo, S.A. French, R.S. Balaban, Am J Physiol (in press).
A. M. Das, D. A. Harris, Biochem Soc Trans 18: 554-5, (1990).
S. A. French, P. R. Territo, R. S. Balaban, Am J Physiol (1998).
R. S. Balaban, V. K. Mootha, A. Arai, Anal Biochem 237, 274-8 (1996).
H. Rottenberg, J Membr Biol, 81, 127-38 (1984).
Fabiato, A., and F. Fabiato, J. Physiol. (Paris), 75: 463-505 (1979).
|
